Empresas Premium





Gracias a la aplicación de nuevos métodos de aprendizaje automático a la investigación en química cuántica, un grupo de investigadores de la Universidad de Heidelberg (Alemania) han conseguido mejoras significativas en química computacional.
Por ello, han logrado dar respuesta a la resolución de un dilema que venía pesando décadas en el campo de la química cuántica: el cálculo preciso y estable de energías moleculares y densidades electrónicas con un enfoque denominado "sin orbitales", que utiliza una potencia computacional considerablemente menor y, por tanto, permite calcular moléculas muy grandes.
En el marco del Cluster de Excelencia STRUCTURES, dos equipos de investigación del Centro Interdisciplinar de Computación Científica (IWR) han perfeccionado un proceso informático considerado poco fiable durante mucho tiempo, de modo que ofrece resultados precisos y establece con fiabilidad una solución físicamente significativa.
La distribución de los electrones en una molécula determina sus propiedades químicas, desde su estabilidad y reactividad hasta sus efectos biológicos. Calcular con fiabilidad esta distribución de electrones y la energía resultante es una de las funciones centrales de la química cuántica.
Estos cálculos constituyen la base de muchas aplicaciones en las que las moléculas deben entenderse y diseñarse específicamente, como por ejemplo para obtener nuevos fármacos, mejores baterías, materiales para la conversión de energía o catalizadores más eficientes. Sin embargo, estos cálculos requieren un gran esfuerzo computacional y se vuelven muy complejos.
Cuanto más grande es la molécula o más variantes hay que comprobar, antes alcanzan sus límites los procesos informáticos establecidos. El proyecto "Química cuántica sin orbitales" se sitúa aquí en la interfaz de la química, la física y la investigación en IA.
En química cuántica, las moléculas se describen a menudo mediante la teoría del funcional de la densidad, que permite predecir las propiedades químicas moleculares sin tener que calcular la función de onda de la mecánica cuántica.
En su lugar, se utiliza la densidad de electrones como magnitud principal, una simplificación que por fin permite realizar cálculos. Este enfoque sin orbitales promete cálculos especialmente eficientes, pero hasta ahora se consideraba escasamente útil, ya que pequeñas desviaciones en la densidad electrónica conducían a resultados inestables o "no físicos". Con la ayuda del aprendizaje automático, el método de Heidelberg resuelve por fin este problema de precisión y estabilidad para muchas moléculas orgánicas diferentes.
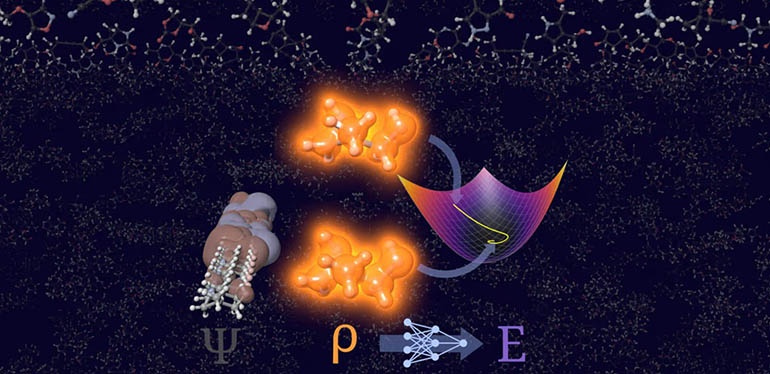
El nuevo proceso, denominado STRUCTURES25, se basa en una red neuronal específicamente desarrollada que aprende la relación entre densidad electrónica y energía directamente a partir de cálculos de referencia precisos, capturando el entorno químico de cada átomo individual en una representación matemáticamente detallada.
Un concepto de entrenamiento único fue fundamental: el modelo se entrenó no sólo con densidades electrónicas convergentes, sino también con muchas variantes en torno a la solución correcta, generadas por cambios específicos y controlados en los cálculos de referencia subyacentes. Por tanto, este proceso informático es capaz de encontrar de forma fiable una solución físicamente significativa para las energías moleculares y las densidades electrónicas, incluso en caso de pequeñas desviaciones. Permanece estable sin "perderse" en el cálculo, subrayan los investigadores de Heidelberg.
En las pruebas realizadas con una amplia y diversa colección de moléculas orgánicas, STRUCTURES25 alcanzó una precisión que puede competir con los cálculos de referencia establecidos, demostrando por primera vez una convergencia estable utilizando un enfoque sin orbitales.
El rendimiento del método se demostró no sólo en ejemplos pequeños, sino también en moléculas "tipo fármaco" considerablemente mayores. Las primeras comparaciones del tiempo de ejecución demuestran que el proceso informático puede escalar mejor con el aumento del tamaño de la molécula y, por tanto, aumentar la velocidad del cálculo. Ahora es posible realizar cálculos que antes se consideraban demasiado complejos.
"Hace tiempo que la teoría funcional de la densidad sin orbitales prometía cálculos más rápidos, pero no a expensas de la física", afirma el Prof. Dr. Fred Hamprecht, que dirige el grupo de investigación "Inteligencia Artificial Científica" del IWR. "Con STRUCTURES25, demostramos por primera vez que la computación puede incluir ambas cosas: energías químicamente precisas y una optimización estable y práctica de la densidad de electrones".
El Prof. Dr. Andreas Dreuw, jefe del grupo de investigación "Química Teórica y Computacional" del IWR, añade: "La optimización ya no es inestable y, por tanto, supone un gran paso adelante para realizar predicciones considerablemente más rápidas con gran precisión. Ahora están al alcance simulaciones que los procesos clásicos apenas podían tocar, como cuando hay que investigar muchas configuraciones o moléculas muy grandes."
La base del trabajo ha sido la estrecha cooperación interdisciplinar de los grupos de investigación del Cluster de Excelencia "STRUCTURES: Un enfoque unificador de los fenómenos emergentes en el mundo físico, las matemáticas y los datos complejos" de la Universidad de Heidelberg. Aquí, investigadores de diversas disciplinas estudian cómo surgen las estructuras, cómo pueden detectarse en grandes conjuntos de datos y las ventajas que ofrecen a la ciencia y la tecnología.
Además del apoyo prestado por el Cluster de Excelencia, la financiación también procedió del programa Wildcard de la Carl-Zeiss-Stiftung, que apoya proyectos especialmente innovadores y audaces. Los resultados de la investigación se publicaron en el "Journal of the American Chemical Society".
|